


Peptide drugs have many advantages over other classes of drugs: they are well tolerated, have stereochemistry that is easy to insert, thus providing selective targets, and can be very effective in interacting with large protein surfaces. In fact, peptides are very good at binding to the considerable and often unstable surfaces on proteins that regulate PPI.
What is Cyclic Peptide?
Cyclic peptides are peptide chains with a cyclic structure, mostly composed of 5-14 amino acids, with a molecular weight of about 500-2000 Da. Cyclic peptides have several advantages over linear peptides. Linear peptides as drugs may be inherently unstable and have a high probability of intracellular proteolysis. Considering that the free acid and free amine are at both ends, they are also usually polar. Cyclization promotes intramolecular hydrogen bonding within the ring structure and reduces the external hydrogen bonding capacity of the molecule, which reduces the polarity of the molecule compared to the acyclic precursor and increases the membrane permeability of the compound. The two β-turn local secondary structures caused by cyclization also reduce polar surface area, which generally enhances cell permeability.
Cyclic peptides are usually produced by end-to-end, cephalic, or side-chain to side-chain cyclization reactions. Introducing cyclization can make the conformation of the peptide chain more stable, thereby increasing the binding affinity to the target protein and reducing nonspecific binding due to fewer alternative conformations. Reduced conformational flexibility reduces the chance of molecules fitting to protease catalytic sites and improves proteomic resistance. Intervention in protein-protein interaction (PPI) by forming larger interaction surfaces to improve peptide chain efficacy.
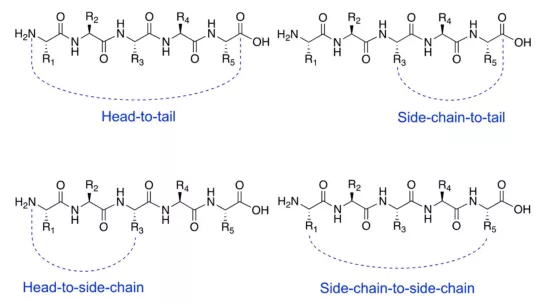
Chem. Eur. J. 2021, 27, 1487-1513
Cell Permeability of Cyclic Peptides
There are three pathways by which cyclic peptides enter cells: passive diffusion pathway, carrier-mediated transport pathway, and endocytosis pathway. The passive diffusion pathway usually requires good amphiphilicity and low molecular weight (<1000Da) of cyclic peptide molecules to enter cells. Some peptide-based drugs are recognized by transporters and subsequently introduced into cells, and some cyclic peptides enter cells through PepT1 and PepT2 transporters. However, these receptors are expressed primarily in the gut (absorption) and kidney (excretion), so they are important for oral bioavailability and elimination of peptide drugs, but are unlikely to be involved in cellular penetration of target tissues. Moreover, the membrane of the endocytic sac is similar to the cell membrane, and non-passive permeable molecules may not be able to effectively escape the endosome and lysosome into the cytoplasm. In order to enhance the cell permeability of peptide drugs, it is necessary to improve the molecular weight and amphiphilicity of drugs. Cyclic peptide drugs, however, are flexible enough to meet these needs. First, controlling the sequence length of the cyclic peptide molecule can control the molecular weight requirements. Secondly, by modifying the side chains of amino acids (introducing unnatural amino acids, forming disulfide bonds, etc.) or main chains (n-methylation, etc.), introducing unnatural hydrophobic groups can enhance the hydrophobicity of cyclic peptide molecules, thus improving the cellular permeability of cyclic peptide drug molecules. Furthermore, the hydrophilic and hydrophobic properties of cyclic peptides can be further optimized according to the symmetry of the framework (d-type amino acids).
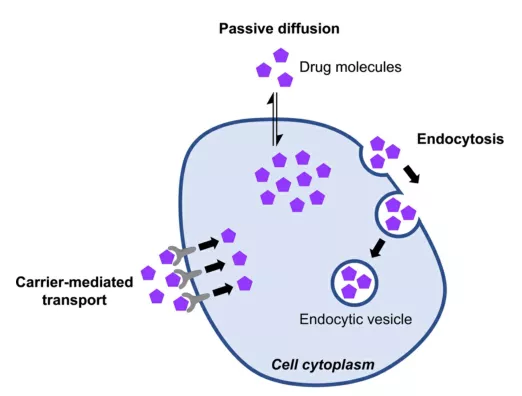
Chem. Eur. J. 2021, 27, 1487-1513
FDA-Approved Cyclic Peptide Drugs
Cyclic peptide drugs targeting intracellular proteins
1.Romidepsin
Romidepsin is a natural bicyclic peptide discovered in Chromobacterium violaceum (1994), containing a head-to-tail lactone cyclization and a pair of disulfide bonds, mainly produced by fermentation. Romidepsin, a histone deacetylase inhibitor (HDACi), was approved by the FDA in 2009 for cutaneous T-cell lymphoma (CTCL) that has received at least one systemic therapy, primarily as an antineoplastic drug. It is a prodrug in which disulfide is reduced to two mercaptols in the intracellular matrix that chelates zinc ions at the zinc-dependent active site of histone deacetylase (HDAC), thereby inhibiting histone deacetylase and inducing apoptosis.
2.Voclosporin
Voclosporin has 11 residues, including 1 D-type amino acid, which is head and tail cyclization. The N-terminus of some amino acids are methylated by S-adenosine methionine. Volcyclosporine was obtained by optimizing cyclosporin A. Voclosporin has better efficacy and metabolic stability than cyclosporin A. As a novel best-in-class calcineurin inhibitor (CNI), voclosporin has a synergistic and dual mechanism of action: it stabilizes renal podocyte by inhibiting CALcineurin (CN), blocking IL-2 expression and T cell-mediated immune response.
Cyclic peptide drugs targeting extramembrane proteins
1.Ziconotide
Ziconotide is a synthetic omega-MVIIA toxin isolated from the venom of Conus Magus. It contains 25 residues, of which six amino acids are cysteine, linked by three pairs of disulfide bonds. As a potent selective blocker of N-type calcium channels, Ziconotide was approved in 2004 for the treatment of severe chronic pain.
2.Linaclotide
Linaclotide is an oral peptide composed of 14 amino acids developed in the 1990s and belongs to the Cyclic Guanosine Monophosphate (cGMP) regulatory peptide family. In 2012, linaclotide was approved by the FDA for the treatment of irritable bowel syndrome with constipation (IBS-C).
3.Plecanatide
Pukanatide is an oral guanylate cyclase C(GC-C) agonist developed by Synergy that regulates acid and base ions in the gastrointestinal tract, inducing fluid transport into the gastrointestinal tract and increasing peristalsis. Pukanatide, a 16-amino acid analogue of uranyin, is produced by replacing Human uranyin Asp3 with Glu3 and binds to guanylate cyclase C(GC-C) receptors at pH5.0 (duodenum and proximal jejunum pH) bound by 2 pairs of disulfide bonds. On 2017-01-19, Plcanatide was first approved by the FDA for the treatment of gastrointestinal disorders such as constipated irritable bowel syndrome (IBS-C) and chronic idiopathic constipation (CIC).
4.Pasireotide
Pasireotide is a second-generation analog of somatostatin, a naturally occurring inhibitory hormone that blocks the release of several other hormones by interacting with the somatostatin receptor (coupled to G protein) , including growth hormone, thyroid-stimulating hormone (TSH), insulin, and glucagon. Compared with other somatostatin analogues, paretide has a 40-fold increase in affinity for somatostatin receptor 5. Pasireotide is a head-to-tail cyclized hexapeptide. Pasireotide has higher binding affinity to the somatostatin receptors SSTR1, SSTR3 and SSTR5 than octreotide and lanreotide.
5.Lanreotide
Lanretide is the first sustained-release, long-acting somatostatin octapeptide analog, which is produced by cyclization of a disulfide bond between cysteine and cysteine, and binds stronger to somatostatin receptors on adenohypophysial cells than octreotide. Studies have shown that lanreotide mainly acts through somatostatin receptor-2 and somatostatin receptor-5, with a strong affinity for peripheral somatostatin receptors and a weaker affinity for central receptors.
6.Vasopressin
Vasopressin, discovered in 1928 and its sequence elucidated in 1951, is an antidiuretic hormone with nine amino acids, cyclized by a disulfide bond between Cys4 and Cys9.
7.Terlipressin
Terisopressin is a synthetic, long-acting vasopressin analogue that acts on the vasopressin receptors V1a, V1b and V2. It is a prodrug that is degraded in vivo by endopeptidase to form lysine-vasopressin by removing the trisglycine residue at the n-terminal of the cyclic peptide, which is the main active metabolite and is further degraded in the liver, kidney and other tissues. As a result, teripressin acts slowly but has a longer duration, with a half-life of 6hr, which is longer than that of vasopressin (10min), and is safer with fewer side effects.
8.Bremelanotide
Bremelanotide is a synthetic peptide molecule consisting of seven amino acids cycled between side chains Asp2 and Lys7 to form the lactam analogue (α-MSH) of alpha-melanocyte-stimulating hormone.
9.Setmelanotide
Setmelanotide is a disulfide cyclized octapeptide that preferentially agonizes the melanocortin 4 receptor (MC4R) with an EC50 of 0.2 nM.
Antimicrobial Cyclic Peptides
1.Daptomycin
Daptomycin is a cyclic lipopeptide antibiotic isolated from Streptomyces rosiana. It contains 13 residues, 2 of which are non-natural amino acids, and 10 of which form lactone rings through the side chain and C-terminal of Thr4.
2.Telavacin
Telavancin is a semi-synthetic vancomycin derivative, which is based on the vancomycin structure. It is obtained by chemical modification to introduce an aliphatic chain on the amino group of the sugar group, and to introduce methylamine methyl phosphate on the seventh aromatic amino acid. Through modification, the binding force with cell membrane and affinity with antibacterial target are enhanced, and teravanin is a fast bactericidal antibiotic for injection.
3.Dalbavancin
Dalbavancin is a second-generation semi-synthetic lipoglycopeptide antibiotic that is more potent than vancomycin and inhibits bacterial cell wall synthesis. The vancomycin family of natural antimicrobial glycopeptides are highly modifiable heptapeptides in which 5 amino acids remain unchanged, while amino acids 1 and 3 are highly differentiated. The heptapeptide core is used to bind the C-terminal D-Ala-D-Ala of the peptidoglycan chain, and the side chain functional groups are derived to improve the properties of the peptide molecule.
4.Oritavancin
Oritavancin, a second-generation, semi-synthetic lipoglycopeptide, is derived from chloroeremomycin, an analogue of vancomycin. Similar to vancomycin, it contains a core heptapeptide.
5.Caspofungin, Micafungin and Anidulafungin
Capofungin, micafengin, and anifengin all belong to the cyclic hexaliphatic peptide family. They share the same cyclic peptide core structure and are cyclized by amidation between the n-end of the side chain of the first lysine residue and the c-end of the peptide chain, and all target the 1,3-β -glucan synthase in the cell wall for the synthesis of glucan. All three antifungal agents have clinical efficacy against invasive candidiasis and other forms of systemic mycosis.
References
Cyclic peptide drugs approved in the last two decades (2001-2021)
Comments