


As a tumor suppressor, p53 is one of the most commonly mutated genes in human cancers. Due to its important role in tumor suppression, p53 has attracted great interest from researchers in drug development. As a transcription factor, p53 is directly or indirectly involved in regulating many genes, including cell cycle arrest, apoptosis, senescence, and DNA repair. In recent years, p53 has become one of the most important and attractive drug targets in cancer treatment. This article will review research on p53-based gene therapy, targeted therapy and immunotherapy.
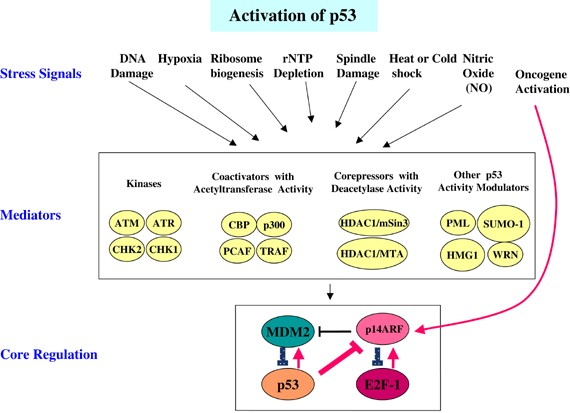
Figure 1. The p53 pathway.
Overview of the Structure and Function of p53 Gene
The human p53 gene is located on chromosome 17p13.1, contains 11 exons, encodes 393 amino acids, and has a relative molecular mass of 43,700. p53 is a sequence-specific DNA-binding protein with two transactivation domains at its N-terminus, followed by a conserved proline-rich domain, a central DNA-binding domain, and finally a The C terminus encodes its nuclear localization signal and an oligomerization domain required for transcriptional activity. p53 mutations or deletions occur in about half of cancer patients, and it is one of the most commonly mutated genes in cancer. p53 mutations mainly occur in the DNA-binding domain, and several amino acid residues have a much higher mutation frequency than other amino acids. These residues are often called hotspot TP53 mutations, among which R175, G245, R248, R249, R273, and R282 are the major hot spot.
As a tumor suppressor, p53 can regulate cell division, prevent cells with mutated or damaged DNA from dividing, and transmit apoptosis signals to these cells, thereby preventing tumor formation. p53 can respond to cellular stress or DNA damage, activate a variety of transcription targets, and coordinate a variety of responses, including cell cycle arrest, DNA repair, antioxidant effects, anti-angiogenesis effects, metabolism, autophagy, senescence, and apoptosis. Under non-stress conditions, p53 is ubiquitinated by E3 proteins such as mouse two-microbody protein 2 (MDM2), constitutive photomorphogen 1 (COP1), RING domain family member 2 (PIRH2), and triple motif protein 24 (TRIM24), and subsequent degradation by the proteasome maintains p53 protein at a low level. Another important negative regulator of p53 is mouse double microbody protein X(MDM4) can inhibit cell cycle progression in a variety of ways, one of which is to upregulate p21 protein expression. Subsequently, p21 protein can bind to cell cycle proteins, causing cell cycle G1 phase arrest. In addition, p53 can also bind to other target genes, such as 14-3-3σ and cell division cycle protein 25 (CDC25), to arrest the cell cycle in the G2/M phase. In addition, p53 also plays an active role in many different types of DNA repair, including nucleotide excision repair, base excision repair, mismatch repair and non-homologous end joining. Beyond this, p53 mutations provide a selective advantage to tumor cells, allowing them to circumvent cell cycle checkpoints, avoid apoptosis and senescence, and proliferate under conditions where normal cells cannot proliferate.
Targeting MDM2 and MDMX
MDM2 inhibitors
MDM2 is a major negative regulator of p53. In cancer cells carrying WT p53, MDM2 is often overexpressed through gene amplification or transcriptional upregulation. Therefore, MDM2 is a good target for cancer treatment. Common inhibitors of MDM2 are Nutlin3a and Nutlin-3a-aa, which are cis-imidazole compounds that can induce p53 activation in WT p53 cancer cells but have no effect in mutant p53 (mutp53) cells. Nutlin- 3a-aa is more active than Nutlin-3a against purified wild-type MDM2 and is more effective in increasing p53 levels and releasing the transcription of p53 target genes from MDM2-induced repression.
MDMX inhibitors
Like MDM2, as a negative regulator of p53, MDMX has also attracted great attention from inhibitor developers, but few inhibitors have been discovered so far, and most of them are short peptides that inhibit MDMX activity. To inhibit the interaction between MDMX and p53, the researchers designed a highly specific short peptide (SAH-p53-8) that can activate p53 in MDMX-dependent cancer cells and induce apoptosis in vivo. However, later studies showed that SAH-p53-8 binds very tightly to serum, which limits its entry into tumor cells and further clinical development.
Targeting p53 mutants
Most tumor cells contain p53 gene alterations, including mutations, deletions, and translocations. However, the incidence of p53 gene mutations is higher than deletions and translocations. p53 mutations not only lead to changes in p53 function, but also confer new functions to these mutants, called gain of function (GOFs). To target cancer cells with p53 mutations, several therapies have been developed.
Activate WT p53 Activity
As a tumor suppressor gene, reactivating the activity of WT p53 is a better treatment method. The p53-Y220C mutant is an excellent example of developing mutant p53 therapeutics through protein stabilization. Some studies have found that small molecule compounds PK083 and PK7080, which target the unique surface cracks produced by the Y220C mutation, can bind to the Y220C mutant, restore the wild conformation, and induce Y220C-dependent cell cycle arrest and apoptosis. Studies have shown that the correct folding of p53 requires the presence of zinc, and the lack of zinc in the DNA-binding domain leads to misfolding of the protein. By providing zinc to certain p53 mutants such as R175H, these mutants can restore the conformation and function of WT p53, once again functioning as a tumor suppressor gene.
GOFs that Inhibit Mutant p53
Most p53-based drug development targets WT p53 activity in cancer cells, but there are also attempts to eliminate mutp53 GOFs activity by targeting mutp53 for rapid degradation. Since the introduction of mutp53 GOFs, many mechanisms of mutp53 GOFs have been proposed, such as: BAG5 interacts with mutp53 protein and promotes the accumulation of mutp53 protein, resulting in GOFs promoting cell proliferation, tumor growth, cell migration and chemical resistance. p53 mutants down-regulate solute carrier family member SLC7A11 (solute carrier family 7 member 11) through the epidermal growth factor receptor (EGFR)/extracellular regulated protein kinase (ERK) cycle or through transcriptional regulatory protein 1 (BACH1)-mediated down-regulation, enhances the invasion and metastasis of cancer cells. A general strategy for GOFs targeting mutant p53 is to utilize existing therapeutics that target pathways or genes affected by mutant p53, such as EGFR inhibitors, statins of the metovalerate pathway, and inhibitors of mixed lineage leukemias (MLLs). Furthermore, there is emerging evidence that the interaction between mutp53 and the tumor microenvironment may shape the GOFs of mutant p53. The GOFs of p53 play a key role in the occurrence and development of cancer. A comprehensive understanding of the GOFs of p53 will help provide a better theoretical basis for personalized drug therapy in cancer treatment.
Targeting Mutant p53 Stability
One strategy to target p53 mutants is to reduce their stability. Some p53 mutants are oncogenic, and reduced levels can lead to cancer cell death. Studies have found that the stability of mutant p53 can be enhanced by the heat shock protein (HSPs) family, and the binding of the HSPs complex prevents the degradation of mutant p53 by E3 ubiquitin ligases such as MDM2 and HSP70-interacting protein (CHIP).
Comments